









İçerik
Bourneville yumrulu skleroz
Bu ne ?
Bourneville tuberoskleroz, vücudun farklı bölgelerinde iyi huylu (kanserli olmayan) bir tümörün gelişmesiyle karakterize karmaşık bir genetik hastalıktır. Bu tümörler daha sonra deride, beyinde, böbreklerde ve diğer organ ve dokularda yerleşebilir. Bu patoloji de bireyin gelişiminde ciddi sorunlara neden olabilir. Bununla birlikte, hastalığın klinik belirtileri ve şiddeti hastadan hastaya değişir.
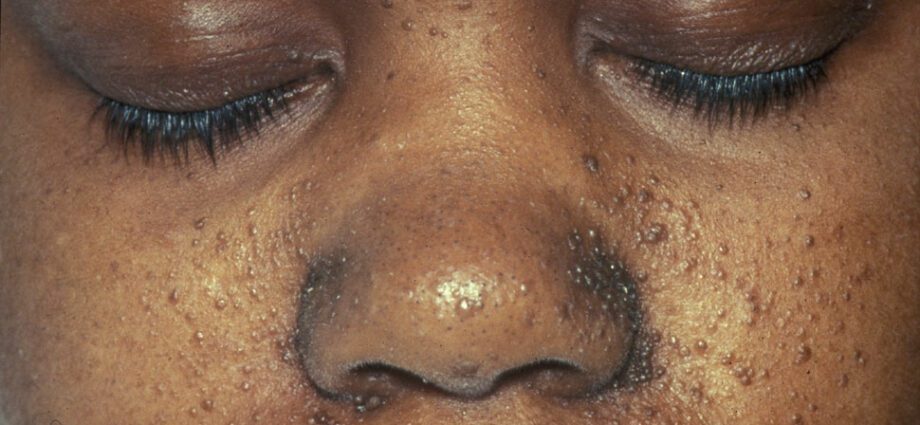
İlişkili cilt anormallikleri genellikle ciltteki lekelere veya cildin vücudun geri kalanından daha açık olduğu bölgelere benzer. Yüzdeki tümörlerin gelişimine anjiyofibrom denir.
Beyin hasarı bağlamında, klinik belirtiler epileptik nöbetler, davranış sorunları (hiperaktivite, saldırganlık, zihinsel engeller, öğrenme sorunları vb.). Hastalığı olan bazı çocuklarda, sosyal etkileşimleri ve iletişimi etkileyen bir tür otizm, gelişimsel bozukluklar bile vardır. İyi huylu beyin tümörleri de hasta için ölümcül olabilecek komplikasyonlara neden olabilir.
Böbreklerde tümör gelişimi, tuberosklerozlu kişilerde yaygındır. Bu, böbrek fonksiyonunda ciddi komplikasyonlara neden olabilir. Ayrıca kalp, akciğer ve retinada tümörler gelişebilir. (2)
Prevalansı (belirli bir zamanda belirli bir popülasyondaki vaka sayısı) 1 / 8 ila 000 / 1 kişi olan nadir bir hastalıktır. (15)
Belirtileri
Bourneville'in tüberosklerozu ile ilişkili klinik belirtiler, etkilenen organlara göre değişir. Ek olarak, hastalıkla ilişkili semptomlar bir kişiden diğerine büyük ölçüde değişir. Hafif ila şiddetli arasında değişen semptomlarla.
Bu hastalığın en yaygın olarak tanımlanan semptomları arasında epileptik nöbetler, bilişsel ve davranışsal bozukluklar, cilt anormallikleri vb. bulunur. En sık etkilenen organlar şunlardır: beyin, kalp, böbrekler, akciğerler ve deri.
Bu hastalıkta kötü huylu (kanserli) tümörlerin gelişmesi mümkündür, ancak bunlar nadirdir ve esas olarak böbrekleri etkiler.
Hastalığın beyindeki klinik belirtileri, farklı seviyelerdeki ataklardan kaynaklanır:
- kortikal tüberküllerde hasar;
– ependimal nodüller (SEN);
– dev ependimal astrositomlar.
Sonuçları: zeka geriliği, öğrenme güçlükleri, davranış bozuklukları, saldırganlık, dikkat bozuklukları, hiperaktivite, obsesif-kompulsif bozukluklar vb.
Böbrek hasarı, kistlerin veya anjiyomiyolipomların gelişimi ile karakterizedir. Bunlar böbrek ağrısına ve hatta böbrek yetmezliğine yol açabilir. Ağır kanama fark edilirse, şiddetli anemi veya yüksek tansiyon olabilir. Özellikle karsinomların (epiteli oluşturan hücrelerin tümörü) gelişimi gibi diğer daha ciddi ancak nadir sonuçlar da görülebilir.
Göz hasarı, retinadaki görünür noktalara benzer şekilde görme bozukluklarına ve hatta körlüğe neden olabilir.
Cilt anormallikleri çoktur:
- hipomelanik maküller: cilde renk veren bir protein olan melanin eksikliğinin bir sonucu olarak, vücudun herhangi bir yerinde ciltte hafif lekelerin ortaya çıkmasına neden olan;
- yüzdeki kırmızı lekelerin görünümü;
- alında renksiz yamalar;
– bir kişiden diğerine bağlı olan diğer cilt anormallikleri.
Hastaların 1/3'ünde hafif kadın baskınlığı olan akciğer lezyonları mevcuttur. İlişkili semptomlar daha sonra az ya da çok şiddetli nefes alma güçlükleridir.
Hastalığın kökenleri
Hastalığın kökeni genetik ve kalıtsaldır.
İletim, TSC1 ve TSC2 genlerindeki mutasyonları içerir. Proteinlerin oluşumunda ilgili genler devreye girer: hamartin ve tuberin. Bu iki protein, etkileşimli bir oyun aracılığıyla hücre çoğalmasını düzenlemeyi mümkün kılar.
Hastalığı olan hastalar, her hücresinde bu genlerin en az bir mutasyona uğramış kopyasıyla doğarlar. Bu mutasyonlar daha sonra hamartin veya tübertin oluşumunu sınırlar.
Genin iki kopyasının mutasyona uğraması durumunda bu iki proteinin üretimini tamamen engellerler. Dolayısıyla bu protein eksikliği vücudun belirli hücrelerin büyümesini düzenlemesine artık izin vermemekte ve bu anlamda farklı doku ve/veya organlarda tümör hücrelerinin gelişmesine yol açmaktadır.
Risk faktörleri
Böyle bir patolojiyi geliştirmek için risk faktörleri genetiktir.
Gerçekten de, hastalığın bulaşması otozomal dominant mod yoluyla etkilidir. Ya, ilgili mutasyona uğramış gen, cinsel olmayan bir kromozomda bulunur. Ayrıca mutasyona uğramış genin iki kopyasından sadece birinin bulunması hastalığın gelişmesi için yeterlidir.
Bu anlamda, hastalıktan muzdarip bu iki ebeveynden birine sahip olan bir bireyin, hasta fenotipini geliştirme riski %50'dir.
Önleme ve tedavi
Hastalığın teşhisi her şeyden önce ayırıcıdır. Atipik fiziksel kriterlere dayanmaktadır. Çoğu durumda, hastalığın ilk karakteristik belirtileri şunlardır: tekrarlayan epileptik nöbetlerin varlığı ve deneğin gelişiminde gecikmeler. Diğer durumlarda, bu ilk belirtiler cilt lekelerine veya bir kalp tümörünün tanımlanmasına neden olur.
Bu ilk tanının ardından tanıyı doğrulamak veya doğrulamamak için ek tetkikler gereklidir. Bunlar şunları içerir:
- beyin taraması;
- beynin MRI'sı (Manyetik Rezonans Görüntüleme);
- kalp, karaciğer ve böbreklerin ultrasonu.
Teşhis, çocuğun doğumunda etkili olabilir. Aksi takdirde, hastanın en kısa sürede sorumluluğunu alması için mümkün olan en kısa sürede yapılması önemlidir.
Şu anda, hastalığın tedavisi yoktur. Bu nedenle ilişkili tedaviler, her bir birey tarafından sunulan semptomlardan bağımsızdır.
Genellikle nöbetleri sınırlamak için anti-epileptik ilaçlar verilir. Ek olarak, beyin ve böbreklerin tümör hücrelerinin tedavisi için ilaçlar da reçete edilir. Davranış sorunları bağlamında, çocuğa özel tedavi gereklidir.
Hastalığın tedavisi genellikle uzun sürelidir. (1)